

Evaluation on 20 Lentinula edodes Strains Using ISSR and F-MSAP Markers
-
摘要: 利用简单序列重复区间扩增多态性(inter-simple sequence repeat,ISSR)和荧光标记甲基化敏感扩增多态性(fluorescence-labeled methylation-sensitive amplified polymorphism,F-MSAP)分子标记技术对收集到的20株香菇Lentinula edodes菌株进行遗传及甲基化多样性分析。ISSR结果表明,8对引物共可扩增出98条稳定清晰可辨的条带,其中多态性条带89条,多态性比率为90.82%,样品间的遗传相似系数范围为0.520 4~0.989 7。F-MSAP结果表明,15对引物共扩增产生13 487个CCGG位点。在全部检测位点中,半甲基化位点为1 704个,平均半甲基化率12.6%;全甲基化位点为1 865个,平均全甲基化率13.8%。进一步将MSAP数据分为甲基化敏感多态性(methylation sensitive polymorphism,MSP)和甲基化不敏感多态性(methylation insensitive polymorphism,MISP)。Mantel检测结果表明,ISSR与MISP分子标记的分析结果有显著的相关性,但ISSR与MSP分子标记的分析结果无显著相关性。说明香菇不同株系间广泛存在DNA甲基化多样性,在育种中具有重要的应用价值。Abstract: The inter-simple sequence repeat (ISSR) and fluorescence-labeled methylation-sensitive amplified polymorphism (F-MSAP) markers were used to evaluate the genetic diversity of 20 Lentinula edodes strains. Ninety-eight bands were detected by 8 ISSR primers, of which 90.82% were polymorphic. The coefficient of pairwise genetic similarity ranged from 0.520 4 to 0.989 7.A total of 13 487 CCGG sites were detected using 15 selected primer pairs. Among the sites, 1 704 were of hemi-methylation with an average rate of 12.63%, and 1 865 of full-methylation with an average rate of 13.83%. The MSAP data were further divided into methylation sensitive polymorphisms (MSP) and methylation insensitive polymorphisms (MISP) groups. A significant correlation between ISSR and MISP was found by the Mantel test, while none between ISSR and MSP. It appeared that there was a significant methylation diversity among various L. edodes which could be of value for the breeding purpose.
-
Keywords:
- Lentinula edodes /
- ISSR /
- methylation /
- MSAP
-
0. 引言
【研究意义】茶树Camellia sinensis(L.)O. Kuntze是我国重要的无酒精饮料作物[1],在生长过程中容易受到低温、干旱、洪涝等胁迫,从而影响茶树的正常生长发育,降低茶叶的产量和质量。植物通过调节乙烯敏感性来对环境胁迫产生反应。乙烯受体ETR(Ethylene receptor)是乙烯信号转导途径的第一个元件,乙烯受体基因表达对调控乙烯信号传递,从而调节植物生长起着重要作用。因此,鉴定并分析乙烯受体基因,挖掘其相关的功能,以期从分子水平了解乙烯受体在茶树生长过程中应对胁迫的作用机制。【前人研究进展】乙烯是通过其与受体的结合来被植物感知的。受体位于细胞内质网膜上,起着负调控乙烯反应的作用[2]。目前,植物乙烯信号转导通路探明:在CU+的参与下,乙烯分子结合乙烯受体,致使CTR1(Constitutive triple response1),失活而减弱下游的EIN2(Ethylene Insensitive2)蛋白的磷酸化,EIN2蛋白c端被切割,c端移动到细胞核使其中的EIN3(Ethylene insensitive3)等因子感受到乙烯信号,促进下游的ERFs(Ethylene response factors)表达,激活乙烯反应[3]。在模式植物拟南芥中,研究者们发现了5种不同类型的乙烯受体,分别为:ETR1、ERS1、EIN4、ETR2和ERS2。每个受体都包含有N端跨膜区、GAF区和组氨酸(His)激酶结构域,ETR1、EIN4和ETR2受体中还包含一个接收域[4]。根据His激酶结构域中是否存在保守元素,将受体分为2个亚科:亚科1(ETR1和ERS1)和亚科2(ETR2、ERS2和EIN4)。前人已在对香蕉[5]、芒果[6]、木薯[7]、丹参[8]等的研究中证实了乙烯受体在果实成熟和花生长等组织生长发育方面的作用以及对逆境胁迫的抵御功能。茶树基因组学的巨大进步有效促进了研究人员对茶叶品质及适制性的理解,2017年云抗10号茶树基因组发表,开启了茶树基因组学研究序幕[9],2018年中国小叶种茶树舒茶早完成测序,舒茶早采用二代测序和三代PacBio序列补洞的策略获得了更高质量的茶树基因组[10]。科学家们已经分离出2个茶叶品种:中国小叶种茶树CSS(Camellia sinensis vars. sinensis)[11]和中国大叶种茶树CSA(Camellia sinensis vars. assamica)[9]的基因组序列,尤其是3个中国种(舒茶早[12]、碧云[13]和龙井43[11])的高精度染色体水平基因组序列已经被科学家们研究获得。但至今鲜有有关茶树乙烯受体基因方面的研究报告。【本研究切入点】前人研究结果表明乙烯受体基因对植物生长发育有内部调控作用。但至今鲜有茶树乙烯受体基因的有关研究报道。【拟解决的关键问题】本研究通过对茶树ETR基因家族进行生物信息学分析,预测其潜在分子功能,明确乙烯受体在茶树发育过程中的动态变化,初步了解家族各成员功能,筛选主要参与茶树组织发育过程ETR基因家族成员,利用前期基因家族的研究及qRT-PCR技术研究在非生物和外源激素胁迫下ETR基因家族各成员相对表达量变化,为进一步探明茶树乙烯受体各成员功能奠定基础。
1. 材料与方法
1.1 供试材料
试验所用材料于福建农林大学茶学福建省高校重点实验室采集。供试原料为相同栽培条件下生长状态良好的二年生铁观音盆栽茶树。原料于2019年4月参照陈丹(2017)等[14]的方法进行了以下处理:
1)低温处理:将在适宜温度(24±2) ℃下生长的原料植株移到人工气候箱中发育,温度设置为4 ℃,湿度为60%~70%,光照时间14 h,黑暗时间10 h。取低温处理后0、4、24、48 h的顶端第2、3片茶树成熟混合叶。
2)植物生长调节剂处理:制备新鲜的GA、MeJA和ABA溶液,提取100 μmol·L−1喷洒于不同植株[15],取各处理后0、6、24、48 h的顶端第2、3片茶树成熟混合叶。
将以上试验材料每个处理取3次重复,用锡纸包裹标记,液氮速冻后存于−80 ℃冰箱中。
1.2 试验方法
1.2.1 CsETR基因家族成员鉴定
首先在茶树基因组数据库TPIA(http://tpia.teaplant.org)[16]中下载茶树ETR基因蛋白序列,使用SMART(http://smart.embl-heidelberg.de/)[17]和CDD(https://www.ncbi.nlm.nih.gov/cdd/)[18]在线网站查找鉴定到的潜在基因的ETR保守结构域,与完整的ETR基因结构域进行比对,以筛选准确的基因家族成员。采用ExPASY(http://www.expasy.org/)[19]和WoLF PSORT(https://wolfpsort.hgc.jp/)[20]在线网站分析CsETR家族成员的蛋白质理化性质并预测其亚细胞定位。
1.2.2 CsETR基因家族系统发育树构建
采用MEGA7.0软件对CsETR基因进行保守序列分析,并于TAIR(https://www.arabidopsis.org/)网站和NCBI(https://www.ncbi.nlm.nih.gov/genbank/)网站下载拟南芥和番茄的乙烯受体基因序列,使用MEGA7.0软件构建系统进化树[21]。
1.2.3 CsETR基因成员结构和保守结构域分析
从TPIA数据库中下载CsETR基因的外显子和内含子数据文件,利用GSDS(http://gsds.cbi.pku.edu.cn/)[22]在线网站制作CsETR家族成员的基因结构图,包含外显子和内含子数目。使用MEME5.0.2(http://meme-suit.org/tools/meme)在线软件分析CsETR基因蛋白序列的保守基序[23]。
1.2.4 CsETR基因的顺式作用元件分析
从TPIA数据库中提取CsETR基因起始位置上游1000 bp序列,利用Editra软件查找启动子顺式作用元件,并于TBtools软件进行分析[24]。
1.2.5 实时荧光定量PCR分析
茶树叶片总RNA使用北京天根多糖多酚植物总RNA提取试剂盒提取,参照北京全式金试剂盒合成茶树乙烯受体cDNA用于qRT-PCR[10]。CsETR家族成员引物设计在primer3plus(http://www.primer3plus.com/)在线网站上进行(表1),内参基因选用CsGAPDH(登录号GE651107)。qRT-PCR反应在荧光定量PCR仪上进行。反应体系及程序参照实验室前期方法,使用2−ΔΔCt方法计算基因相对表达量[25],并使用TBtools绘制热图。
表 1 茶树CsETR家族成员引物序列Table 1. Primer sequence of CsETRs in tea plants基因名称
Gene name上游引物
Forward primers
(5′→3′)下游引物
Reverse primer
(5′→3′)ERS1-1 AATGGCCTGCTGATGAGCTT CACAGAAGATCCAGCAGGCA ERS1-2 GAGGATGGCAGCCTTCAACT AGTACATCCCTTGCCAAGCC ERS1-3 GGCACCAGATTTACCTGCCT GCTGAGCTTTCTGCAACACC EIN4 AATTGTGCCGTTTGGATGCC GAGTTTCTTGCCTGGCTTGC ETR2-1 GCAAATCGCTTGATCGGCAT CCATCATTGCGCTCTGCTTC ETR2-2 ATAAAGGAAGCCGCTTGCCT TCCCGGTTCAGAGATGCCTA CsGAPDH TTGGCATCGTTGAGGGTCT CAGTGGGAACAGGAAAGC 2. 结果与分析
2.1 CsETR基因家族成员的鉴定
为准确鉴定CsETR家族成员,本研究采用了Blast比对方法,并将鉴定到的潜在CsETR家族成员提交至SMART和CDD网站再次进行鉴定,最终获得6个CsETR家族成员,分别命名为CsERS1-1、CsERS1-2、CsERS1-3、CsEIN4、CsETR2-1、CsETR2-2(图1)。采用ExPASY和WoLF PSORT在线网站分析6个家族成员各项因子的理化性质(表2)。通过蛋白质理化分析后得出结果,CsETR的ORF氨基酸长度为614 aa(CsERS1-1)~763 aa(CsETR2-1),分子量为68.63~85.56 kD,等电点大小为6.09(CsERS1-1)~7.57(CsETR2-2),亲水性为0.014~0.250,表明CsETR基因蛋白为碱性疏水蛋白;通过亚细胞定位预测分析后发现,CsETR基因蛋白均位于质膜上。
表 2 茶树CsETR基因家族的序列特征Table 2. Characteristics of CsETR sequences in tea plants基因
Gene name基因组编号
Genome ID编码序列长度
CDS/bp开放阅读框长度
ORF/aa分子量
MW/kD等电点
pI平均亲水性
Grand average of hydropathicity亚细胞定位
Subcellular localizationCsERS1-1 TEA017521 1845 614 68.63 6.09 0.162 质膜 Plasma membrane CsERS1-2 TEA032252 1923 640 71.71 7.02 0.144 质膜 Plasma membrane CsERS1-3 TEA012203 1932 643 71.89 6.67 0.250 质膜 Plasma membrane CsEIN4 TEA025082 2292 763 85.56 7.27 0.014 质膜 Plasma membrane CsETR2-1 TEA002824 2292 763 84.73 6.32 0.077 质膜 Plasma membrane CsETR2-2 TEA020178 2292 763 85.56 7.57 0.054 质膜 Plasma membrane 2.2 CsETR家族成员的系统发育和分类分析
为了解ETR蛋白之间的进化关系,本研究选取了3个典型物种的17个ETR蛋白用来构建系统进化树。共采用6个茶树乙烯受体基因、6个番茄乙烯受体基因和5个拟南芥乙烯受体基因来构建系统发育树(图2)。ETR家族总共可分为2个亚家族,分别为ETR家族和ERS家族。另外,番茄的变异受体基因Nr单独形成一组,与其他2个亚家族功能有所区别。
![]() 图 2 茶树与其他植物蛋白的ETR基因系统进化树注:Cs.茶树;At.拟南芥;Le.番茄。Figure 2. Phylogenetic tree of ETRs in tea and other plant proteinsNote: Cs: Camellia sinensis; At: Arabidopsis thaliana; Le: Lycopersicon esculentum.
图 2 茶树与其他植物蛋白的ETR基因系统进化树注:Cs.茶树;At.拟南芥;Le.番茄。Figure 2. Phylogenetic tree of ETRs in tea and other plant proteinsNote: Cs: Camellia sinensis; At: Arabidopsis thaliana; Le: Lycopersicon esculentum.2.3 CsETR家族成员结构和保守结构域分析
为分析CsETR基因家族成员结构的多样性,本文通过在GSDS网站上制作基因结构图来比较乙烯受体家族的内含子/外显子数量(图3),通过比较这6个成员的基因结构发现,外显子的数量从1(ERS1-2)至12(ERS1-3)个不等,差别较大,其中ERS1-3基因序列最长,超过了10000 bp,ERS1-2除外显子外,无内含子的存在,其他成员内含子均被外显子中断隔开。除此之外,发现ETR亚家族的基因序列分布相似,外显子内含子数量均为2个,而ERS亚家族基因序列分布不均。
![]() 图 3 CsETR基因家族结构注:a.CsETR家族系统进化树;b.CsETR基因结构。Figure 3. Structure of CsETR familyNote: a: phylogenetic tree of CsETR family; b: structure of CsETR.
图 3 CsETR基因家族结构注:a.CsETR家族系统进化树;b.CsETR基因结构。Figure 3. Structure of CsETR familyNote: a: phylogenetic tree of CsETR family; b: structure of CsETR.利用分析网站MEME预测发现了ETR家族的6个保守基序(图4)和基因的保守基序分布及长度,结果显示所有家族成员都有相同的保守基序,且2个亚家族之间的分布在分布区域及长度上都十分相似。说明ETR家族成员基因功能较为保守,2个亚家族分别有着相似的遗传功能。
![]() 图 4 CsETR基因的保守基序分析注:a.CsETR家族系统进化树;b.CsETR家族保守基序分析;c.基序logo分析。Figure 4. Conservative motif analysis on CsETRsNote: a: phylogenetic tree of CsETR family; b: conserved motif distribution of CsETR family; c: motif logo analysis.
图 4 CsETR基因的保守基序分析注:a.CsETR家族系统进化树;b.CsETR家族保守基序分析;c.基序logo分析。Figure 4. Conservative motif analysis on CsETRsNote: a: phylogenetic tree of CsETR family; b: conserved motif distribution of CsETR family; c: motif logo analysis.2.4 CsETR基因家族启动子顺势元件分析
启动子顺势元件对于调控基因表达发挥着重要作用。CsETR基因启动子顺式作用元件使用Plant-CARE在线工具分析(图5)。最终获得3类顺势元件,与茶树生长相关的顺势元件:玉米醇溶蛋白代谢调控(O2-site,1个)、光响应(TCT-motif,25个);胁迫相应元件:厌氧响应(ARE,10个)、干旱响应(MBS,3个)、低温响应(LTR,3个)、防御和压力响应(TC-rich repeats,1个)、创伤响应(WUN-motif,1个)以及激素响应元件:包括生长素(AuxRR-core,1个)、脱落酸(ABmotif,1个)以及激素响应元件:包括生长素(AuxRR-TATbox、GARE-motif,3个)、水杨酸(TCA-element,1个)。由图可知,在ETR家族中6个成员均含有大量顺势元件,其中CsETR2-1含有1个O2-site,CsETR1-3含有1个AuxRR-core,CsETR2-2含有1个WUN-motif。以上结果表明,CsETR家族在茶树的生长发育、激素和外界胁迫响应中起着重要作用。
2.5 CsETR家族成员在不同组织中的表达模式
为研究CsETR基因家族对茶树组织生长发育的功能,从TPIA数据库中下载8种茶树组织表达数据,利用TBtools软件分析其在不同组织中的表达模式(图6)。根据结果可分析出:ETR家族在果实中的表达最为明显,茎和顶芽次之,在根和老叶部分表达相对较低,表明CsETR家族对茶树的果实发育起着重要作用。分析各个基因家族成员的组织表达量发现,CsETR家族有着较为明显的组织表达特异性,其中CsERS1-1、CsEIN4和CsETR2-2基因在果实中表达量较高,说明这3个基因在乙烯对果实成熟的影响过程中扮演着重要角色,而CsERS1-2、CsERS1-1在茎组织有较高表达,CsETR2-2在成熟叶中表达量较低。
![]() 图 6 CsETR基因家族的组织表达谱注:蓝色代表低表达,橙色代表高表达。Figure 6. Expression patterns of CsETR familyNote: blue represents low expression, and red high expression.
图 6 CsETR基因家族的组织表达谱注:蓝色代表低表达,橙色代表高表达。Figure 6. Expression patterns of CsETR familyNote: blue represents low expression, and red high expression.2.6 CsETR家族在4种处理下的表达模式
为探究CsETR基因家族在激素和胁迫下的生理反应,鉴定外界胁迫和激素影响下的乙烯受体表达,本研究使用荧光定量技术测定了CsETR家族6个成员在低温(4 ℃)、赤霉素、脱落酸、茉莉酸处理下的相对表达量(图7)。在对低温4 ℃诱导下的试验材料进行PCR分析后得出其不同时段的基因表达量,发现CsETR基因家族的6个成员表达量均呈现上调趋势,其中CsERS1-1表达量上调幅度最大,CsERS1-3较小,大部分基因在低温诱导48 h后的表达量最高。分别对GA、MeJA、ABA处理后试验材料基因表达量进行分析,在GA的影响下,除去EIN4基因的表达量略有波动外,大部分ETR基因家族成员表达量均呈上调趋势,其中CsERS1-3的上调幅度最大,而CsERS1-1在6~48 h的基因表达量差别不大。在JA影响下的ETR基因表达量均呈现上调趋势,且CsERS1-3的上调幅度亦位于首位。在ABA的影响下,除CsETR2-2基因的表达量呈下调趋势外,其他家族成员都呈上调趋势,其中CsERS1-3的上调幅度依旧稳居首位。然而,ETR基因家族成员在ABA处理6 h后的表达量变化均难以测定,但内参处理下的基因表达量变化正常,推测其原因是ABA处理6 h对CsETR基因的影响不明显。
![]() 图 7 CsETR基因在低温、GA、MeJA、ABA处理下的表达谱注:对同一个基因相同处理下不同时间点的表达量进行差异性分析( “*”表示P<0.05)Figure 7. Expression profile of CsETRs under low-temperature, GA, MeJA, or ABA treatmentNote: Expressions of same gene under identical treatment at different time ("*" indicates P< 0.05).
图 7 CsETR基因在低温、GA、MeJA、ABA处理下的表达谱注:对同一个基因相同处理下不同时间点的表达量进行差异性分析( “*”表示P<0.05)Figure 7. Expression profile of CsETRs under low-temperature, GA, MeJA, or ABA treatmentNote: Expressions of same gene under identical treatment at different time ("*" indicates P< 0.05).3. 讨论与结论
前人研究表明,ETR基因对植物生长,尤其是在成花及果实成熟阶段发挥着重要的调节作用。目前已经在拟南芥中鉴定到5个ETR成员[10]、番茄中鉴定出6个ETR成员[26]、甜瓜中鉴定出3个家族成员[27]、水稻中分离出5个基因[28]、烟草中鉴定出4个ETR家族成员[29]。在对ETR基因的进化史研究发现,ETR家族成员中,类ETR1受体首先进化,可能伴随着茶藻进化过程。乙烯反应在从藻类向陆地环境过渡期间可能很重要,对乙烯应对外界环境的进化研究有贡献[30]。然而,目前尚无对茶树ETR家族的研究。本研究在茶树基因组数据的基础上,根据其蛋白质结构鉴定出了6个ETR成员,分析结果介于拟南芥和番茄之间。通过对模式植物拟南芥ETR基因家族的分析发现,ETR基因分为2个亚家族,茶树ETR基因在这2个亚家族中均有分布,其中亚家族1中ERS基因有3个。其中,CsETR2-1和CsETR2-2与LeETR4同源性最为接近;CsERS1-1和CsERS1-3与AtERS1同源性最近,表明它们可能行使了相同的功能。这些不同的主要序列,存在区域和表现出激酶活性的类型,可能表明受体间的功能差异性。在分析ETR家族结构之后,发现ETR基因外显子-内含子高度保守,外显子数目为1~12个,同一组间的外显子数目相似,说明同一组基因间有着功能的相似之处。而在MEME网站上分析CsETR蛋白质的基序发现,6个基因都存在相同的6个基序,且相同组的距离几乎相同,说明2个亚家族有着相似的遗传功能。
乙烯受体是乙烯信号转导通路中的第一步,对乙烯引起的植物生理反应有着重要的作用。研究表明,ETR基因对于调节植物果实成熟和花衰老有着关键作用。不同的乙烯受体,即使结构相似,在同一或不同植物中对于植物生长阶段的表达水平与模式都有差异,如在同一株月季中,亚家族1的2个成员RhETR1和RhETR3结构相似,但在切花开放过程中,2个基因的表达却完全相反,RhETR1上调,RhETR3则下调。在2个不同植物番茄和甜瓜中,番茄LeETR1与甜瓜Cm-ETR1同源,在果实成熟时,LeETR1表达不受乙烯合成影响,而Cm-ETR1的表达随内源乙烯的合成同比变化[31]。ETR基因负调控乙烯信号转导,即上调ETR基因的表达,植物的乙烯敏感性就降低,反之升高。因此,可以通过调节ETR基因的表达来调控植物对乙烯的敏感性,如在水稻培育过程中通过上调ETR2基因的表达来降低转基因植株的敏感性,从而延迟水稻开花[32]。本研究发现CsETR家族在茶树果实中高表达,尤其是CsERS1-1、CsEIN4、CsETR2-2,说明这些基因对于调控茶果成熟起着重要作用,其次,ERS亚家族对茶树茎高表达,说明这些基因在茶树茎的发育中发挥作用。
研究基因启动子有助于了解植物基因表达模式的差异及其分子调控机制。前人研究发现,许多ETR基因自身含有响应非生物胁迫的顺式作用元件,如在番茄中SlETR6基因含有LTR、MBS、ARE、TCT-motif、ERF(乙烯应答)等顺式作用元件[33]。甘蔗的受体基因SoERS1中也含有干旱诱导的MYB结合位点和LTR元件,说明该基因的表达可能受干旱和高温等因素的调控[34]。同样,我们在对茶树的启动子研究当中,也发现了每个成员所携带的顺式作用元件,如ARE、TCT-motif、LTR、ABRE、AuxRR-core、CGTCA-motif等,大部分都与非生物胁迫和激素有关。在对低温及植物生长调节剂处理下的茶叶经PCR分析后发现,CsETR家族所有成员在低温胁迫下的表达量均呈上调趋势,这与启动子顺势元件分析结果相匹配。在GA、ABA、JA的诱导下,大部分家族成员基因表达量都呈上调趋势,而EIN4基因在GA处理6 h后达到最高值,说明EIN4基因在GA处理前期正调控反应最大,而ETR2-2基因在ABA处理下表达量呈下调趋势,可能是ETR2-2基因在ABA的影响下表达收到抑制。在CsETR家族基因中,CsERS1-3在植物生长调节剂的影响下表达量上调幅度最大,说明ERS1-3在对植物生长调节剂的响应中可能发挥重要的正调控作用。
本研究首次开展了对茶树全基因组数据库中CsETR基因家族的鉴定分析,鉴定出6个家族成员,分为2个亚科,对其保守结构域、启动子顺势元件分析以及在低温、外源激素胁迫下的反应进行分析后得出初步结论:茶树乙烯受体基因家族与茶树的组织生长发育、非生物胁迫和外源激素影响有着密切的关系,后续还需进行细致的试验具体验证这些基因的生物学功能,并为茶树新品种选育提供参考。
-
![]()
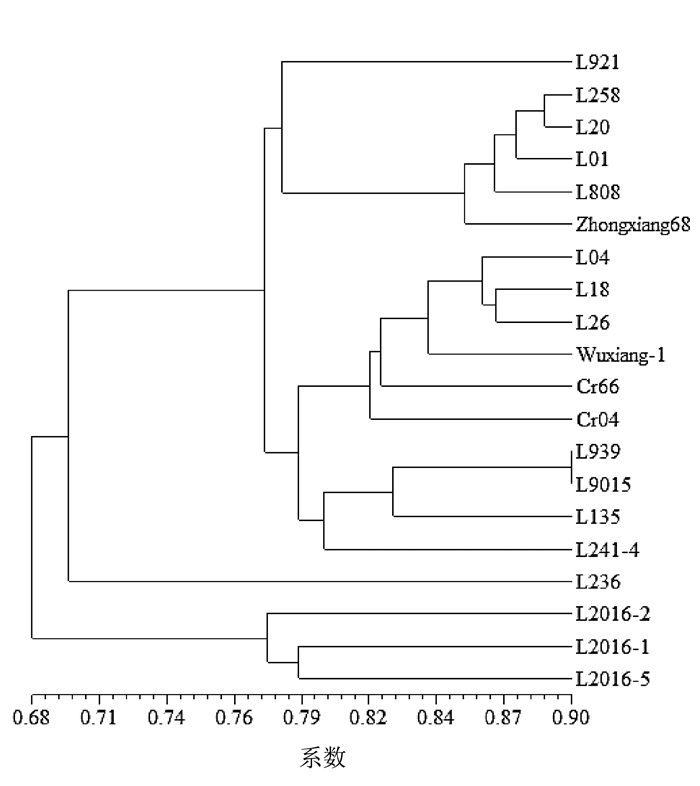
图 1 20株香菇ISSR分析的UPMGA聚类
Figure 1. UPMGA dendrogram based on ISSR data on 20 L. edodes strains
![]()
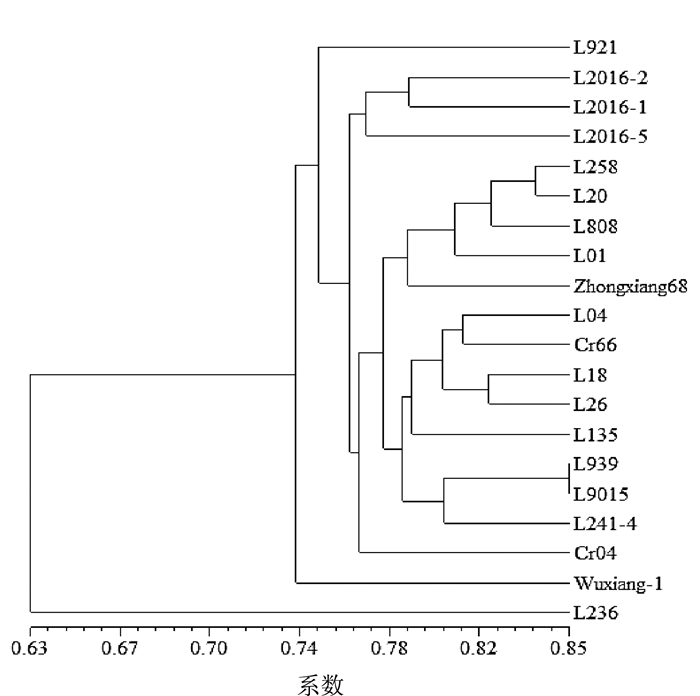
图 2 20株香菇MISP分析的UPMGA聚类
Figure 2. UPMGA dendrogram based on MISP data on 20 L. edodes strains
![]()
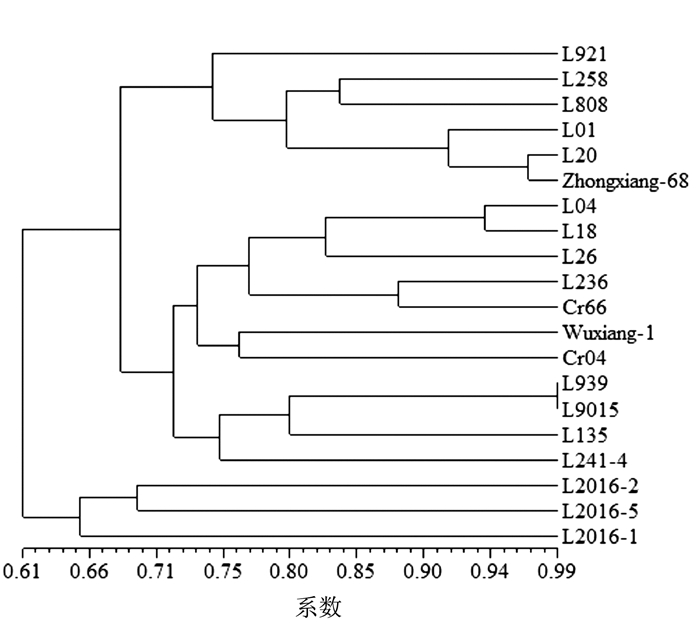
图 3 20株香菇MSP分析的UPMGA聚类
Figure 3. UPMGA dendrogram based on MSP data on 20 L. edodes strains
表 1 20株香菇DNA甲基化水平分析
Table 1 Analysis of methylation polymorphisms on 20 L. edodes strains
菌株 带型 半甲基化率/% 全甲基化率/% 总甲基化率/% Ⅰ Ⅱ Ⅲ Cr04 516 101 70 14.70 10.19 24.89 Cr66 538 64 93 9.21 13.38 22.59 L01 502 78 101 11.45 14.83 26.28 L04 484 85 106 12.59 15.70 28.30 L135 490 78 78 12.07 12.07 24.15 L18 510 91 70 13.56 10.43 23.99 L20 537 76 88 10.84 12.55 23.40 L2016-1 449 77 89 12.52 14.47 26.99 L2016-2 478 100 82 15.15 12.42 27.58 L2016-5 503 108 94 15.32 13.33 28.65 L236 341 78 242 11.80 36.61 48.41 L241-4 537 77 84 11.03 12.03 23.07 L258 497 74 91 11.18 13.75 24.92 L26 549 79 77 11.21 10.92 22.13 L808 504 74 88 11.11 13.21 24.32 L9015 500 89 70 13.51 10.62 24.13 L921 479 81 91 12.44 13.98 26.42 L939 503 97 54 14.83 8.26 23.09 Wuxiang-1 498 91 114 12.94 16.22 29.16 Zhongxiang-68 503 106 83 15.32 11.99 27.31 注:总扩增位点= Ⅰ+Ⅱ+Ⅲ;甲基化总位点= Ⅱ+Ⅲ;总甲基化率=(Ⅱ+Ⅲ)/(Ⅰ+Ⅱ+Ⅲ);全甲基化率=Ⅲ/(Ⅰ+Ⅱ+Ⅲ);半甲基化率= Ⅱ/(Ⅰ+Ⅱ+Ⅲ)。  下载: 导出CSV
下载: 导出CSV
-
[1] ZIETKIEWICZ E, RAFSLSKI A, LABUDA D. Genome fingerprinting by simple sequence repeat(SSR)anchored polymerase chain re-action amplification[J]. Genomics, 1994, 20(2):176-183. DOI: 10.1006/geno.1994.1151
[2] 陆娜, 周祖法, 王伟科, 等.利用ISSR分子标记鉴别杏鲍菇生产菌株的研究[J].北方园艺, 2015, 39(21):150-152. http://d.old.wanfangdata.com.cn/Periodical/bfyany201521038 [3] LIU J, WANG Z R, LI C, et al. Evaluating genetic diversity and constructing core collections of Chinese Lentinula edodes cultivars using ISSR and SRAP markers[J]. Journal of Basic Microbiology, 2015, 55(6):749-760. DOI: 10.1002/jobm.v55.6
[4] 刘晓红. ISSR辅助杏鲍菇常规杂交育种研究[D].合肥: 安徽农业大学, 2010. http://cdmd.cnki.com.cn/Article/CDMD-10364-1011024461.htm [5] LI W, WANG Y, ZHU J, et al. Differential DNA methylation may contribute to temporal and spatial regulation of gene expression and the development of mycelia and conidia in entomopathogenic fungus Metarhizium robertsii[J]. Fungal Biology, 2017, 121(3):293-303. DOI: 10.1016/j.funbio.2017.01.002
[6] 肖冬来, 马璐, 张迪, 等.光照诱导广叶绣球菌基因组甲基化分析[J].食用菌学报, 2017, 24(4):6-11. http://d.old.wanfangdata.com.cn/Periodical/syjxb201704002 [7] PENG H, JIANG G H, ZHANG J, et al. DNA methylation polymorphism and stability in Chinese indica hybrid rice[J]. Science China-Life Sciences, 2013, 56(12):1097-1106. DOI: 10.1007/s11427-013-4576-z
[8] ZHOU H, MA T Y, ZHANG R, et al. Analysis of different ploidy and parent-offspring genomic DNA methylation in the loach Misgurnus anguillicaudatus[J]. International Journal of Molecular Sciences, 2016, 17(8):1299. DOI: 10.3390/ijms17081299
[9] MÖLLER E M, BAHNWEG G, SANDERMANN H, et al. A simple and efficient protocol for isolation of high molecular weight DNA from filamentous fungi, fruit bodies, and infected plant tissues[J]. Nucleic Acids Research, 1992, 20(22):6115. DOI: 10.1093/nar/20.22.6115
[10] YANG T W, MA L H. Extracting DNA from edible fungus by combined method of CTAB and DNA gel purification Kit[J]. Biotechnology, 2009, 19(1):32-34. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=swjs200901012
[11] ZHAO Y, CHEN M Y, STOREY K B, et al. DNA methylation levels analysis in four tissues of sea cucumber Apostichopus japonicus based on fluorescence-labeled methylation-sensitive amplified polymorphism (F-MSAP) during aestivation[J]. Comparative Biochemistry and Physiology B-Biochemistry & Molecular Biology, 2015, 181:26-32.
[12] CERVERA M T, RUIZ-GARCÍA L, MARTÍNEZ-ZAPATER J. Analysis of DNA methylation in, Arabidopsis thaliana, based on methylation-sensitive AFLP markers[J]. Molecular Genetics and Genomics, 2002, 268(4):543-552. DOI: 10.1007/s00438-002-0772-4
[13] 李毅丹.中国松嫩草原短芒野大麦[Hordeum brevisubulatum (Trin.) Link]人工种群的分子遗传与表观遗传多样性及其种群遗传结构的研究[D].长春: 东北师范大学, 2007. [14] ZHONG X F, WANG Y M, LIU X D, et al. DNA methylation polymorphism in annual wild soybean (Glycine soja Sieb. et Zucc.) and cultivated soybean (G. max L. Merr.)[J]. Canadian Journal of Plant Science, 2009, 89(5):851-863. DOI: 10.4141/CJPS08215
[15] FANG J G, SONG C N, QIAN J L, et al. Variation of cytosine methylation in 57 sweet orange cultivars[J]. Acta Physiology Plant, 2010, 32(6):1023-1030. DOI: 10.1007/s11738-010-0491-0
[16] 刘靖宇, 宋秀高, 叶夏, 等.香菇菌株遗传多样性ISSR、RAPD和SRAP综合分析[J].食用菌学报, 2011, 18(3):1-8. DOI: 10.3969/j.issn.1005-9873.2011.03.001 -
期刊类型引用(1)
1. 青亚林,侯炳豪,张宇航,曾珊珊,高婷,叶乃兴. 茶树GATA基因家族的全基因组鉴定及表达分析. 江苏农业科学. 2024(09): 42-50 .  百度学术
百度学术
其他类型引用(0)







计量
- 文章访问数: 898
- HTML全文浏览量: 176
- PDF下载量: 16
- 被引次数: 1
