

Genetic Variations in Southern High-protein Soybean Fudou 234 by Re-sequencing
-
摘要:目的 揭示南方高蛋白大豆遗传变异提供理论参考。方法 通过高通量测序技术对南方高蛋白大豆品种福豆234进行全基因组重测序。结果 共获得
64 757 037 条Clean reads,测序深度为17×,基因组覆盖度分别达98.08%(1×)和96.25%(5×)。共鉴定出1478393 个单核苷酸多态性(Single nucleotide polymorphism, SNP)位点和356739 个小片段插入缺失(Small insertion-deletion, Small InDel)位点。其中共鉴定出14323 个非同义SNP突变的基因,4186 个Small Indel的突变基因位于编码区(Coding sequence, CDS)。通过COG(Clusters of orthologous groups of proteins)分析发现信号传导机制、转录、碳水化合物转输和代谢等和KEGG(Kyoto encyclopedia of genes and genomes)分析发现碳代谢、淀粉和蔗糖代谢、氨基酸生物合成、植物激素信号传导、内质网蛋白质加工等通路与福豆234遗传变异相关。此外,本研究以大豆籽粒蛋白质含量数量性状基因座(Quantitative trait locus, QTL)的两个主要区段内的候选基因进行分析,发现有65个基因发生SNP或Small InDel水平的变异,其中,SNP变异类型达10种,Small InDel变异类型达7种。结论 本研究初步揭示南方高蛋白大豆的遗传变异规律,为高蛋白大豆品种选育和分子标记的开发提供数据支持和理论依据。Abstract:Objective Genetic variations in the southern high-protein soybeans were revealed by re-sequencing the whole genome.Method High-throughput sequencing was conducted on the whole genome of the southern high-protein soybean Fudou 234 to detect genetic variations.Result In the64757037 clean reads, the sequencing depth was 17× with genome coverage of 98.08% (1×) and 96.25% (5×). There were1478393 single nucleotide polymorphisms (SNPs) and356739 small insertion-deletions (Small InDels) identified. Among them,14323 non-synonymous SNP mutations and4186 Small InDel mutated genes were found in the coding sequence (CDS). Analysis by COG (Clusters of orthologous groups of proteins) revealed that signal transduction mechanisms, transcription, carbohydrate translocation and metabolism and KEGG(Kyoto encyclopedia of genes and genomes) analyses revealed that the pathways of carbon metabolism, starch and sucrose metabolism, amino acid biosynthesis, phytohormone signalling, and endoplasmic reticulum protein processing were associated with genetic variation in Fudou 234. In addition, by studying the candidate genes in two major segments of soybean kernel protein quantitative trait locus (QTL), 10 SNP and 7 Small InDel type variations were discovered in 65 genes.Conclusion The genetic variations in the southern high-protein soybeans deviated from the regular varieties were unveiled to provide new venue for breeding and developing molecular markers in studying soybeans.-
Keywords:
- Fudou 234 /
- whole genome re-sequencing /
- single nucleotide polymorphism /
- small InDel /
- variation
-
0. 引言
【研究意义】大豆(Glycine max L. Merr.)是全球重要的粮食作物之一,为食用和饲用提供植物蛋白和油脂来源[1]。随着消费者对大豆需求的增加,育种工作者有望挖掘出大豆优异性状的品种,如高油、高蛋白、抗逆性强、早熟品种等。作为我国传统的经济作物,大豆种质资源保护和利用势在必行。【前人研究进展】近年来,全基因组重测序在分子育种和基因组研究遗传多样性中发展迅速,通过与基因组参考物比对,重测序个体可以识别DNA序列的遗传变异信息,能够有效提升对作物基因组的认识和选择育种提供新见解,因此可以获得更全面的基因组信息,应用于作物改良[2−3]。为发掘大豆优异种质特性、筛选和鉴定影响大豆产量、品质和抗逆性等性状的代谢通路和候选基因提供良好的平台[4]。目前,基于全基因组重测序的大豆优异性状研究成为热点。Yang等[5]对250份大豆进行全基因组重测序,发现GSTT1、GL3、GSTL3、CKX3、CYP85A2等基因参与调控大豆产量和品质性状。Liu等[6]对鲜食大豆进行全基因组重测序技术,发现高质量的SNP大多位于基因间区和内含子,位于编码区较少;同时鉴定出355.55 K InDel,其中
4856 个InDel位于外显子区域。蔗糖合酶基因(SoyZH13_18G042801、SoyZH13_18G042802、SoyZH13_18G042803和SoyZH13_20G077905)、蔗糖磷酸合酶基因(SPS3)和糖转运基因(SoyZH13_04G095700、SoyZH13_09G175600、SWEET6b和SWEET16)作为鲜食大豆甜度和籽粒大小等重要性状相关的候选基因。Lee等[7]基于大豆早花突变体全基因组重测序鉴定了332821 个SNP和65178 个InDel,其中GI、AGL18、TOC1和ELF3等基因影响GmFT2a的表达,导致花期提前。Maldonado dos Santos等[8]对28份巴西大豆通过全基因组重测序鉴定出541762 个SNP、98922 个InDel和1093 个(Copy number variation,CNV),此外还发现共有327个基因的668个等位变异与适应热带气候有关。Jiang等[9]对427个大豆重组自交系进行全基因组重测序,在2号染色体RSC11K区域鉴定出429个SNP和142个InDel特异位点,同时发现34个与大豆抗花叶病毒的相关候选基因。Yuan等[10]对大豆近等基因系进行全基因组重测序,鉴定出55853 个SNPs和3188 个InDel,多态性位点主要富集在13号染色体,还发现候选基因Glyma.13g25950、Glyma.13g25970和Glyma.13g26380与大豆抗花叶病毒有关。【本研究切入点】目前,基于全基因组重测序分析多数聚焦于大豆含油量、甜度、产量和抗逆性等性状,而在大豆高蛋白性状上却鲜有研究。因此,聚焦于全基因组水平上剖析大豆高蛋白性状的遗传多样性及遗传变异具有重要意义。【拟解决的关键问题】本研究旨在对南方高蛋白大豆品种福豆234进行全基因组重测序,对其SNP和Small InDel进行深度挖掘和分析,通过功能富集分析对变异基因进行注释。以期为高蛋白大豆遗传多样性、种质资源鉴定、遗传改良及亲本选配提供理论依据。1. 材料与方法
1.1 植物材料
大豆品种福豆234(国审豆2007024)由福建省农业科学院作物研究所自主选育,为高蛋白品种,蛋白质含量和脂肪含量分别达46.33%和17.68%,适应性广,丰产性和稳产性好[11]。将种子种于蛭石中,待长出三出复叶时,采集三出复叶中间叶片,液氮冻存后于−80 ℃保存备用。
1.2 大豆叶片DNA 提取、文库构建和质量控制
采用改良十六烷基三甲基溴化铵(Hexadecyl trimethyl ammonium bromide, CTAB)法提取大豆叶片基因组DNA[12]。利用Nanodrop 2000检测DNA样品的纯度、浓度和完整性,并用琼脂糖凝胶电泳检测DNA质量。随后将提取的高质量DNA送样北京百迈克生物科技有限公司进行全基因组重测序,在片段大小选择和扩增之前,通过添加腺嘌呤核苷酸(A)和测序适配器来修复DNA,以清除碎片DNA的3'端的间隙,然后通过1%琼脂糖凝胶中电泳进一步选择修复的DNA 片段[13]。通过PCR扩增产物构建测序文库,利用Illumina HiSeqTM
2500 测序平台对合格质量控制的文库进行测序[14]。通过去除测序适配器并过滤低质量reads,对Raw reads进行处理以获得Clean reads。通过BWA(Burrow-Wheeler Aligner)软件[15]将Clean reads映射到参考基因组序列Williams 82 (Glyma.Wm82.a2v1, http://soybase.org)。利用SAMtools软件[16]将对齐后的sam文件转换成bam文件,Picard(http://sourceforge.net/projects/picard.)用于删除标记 PCR 重复项,以此对结果进行分析。1.3 SNP和 Small InDel的注释
基于样本与参考基因组的比对结果,利用GATK软件[17]的HaplotypeCaller算法检测 SNP和 Small InDel。为降低 SNP和 Small InDel检测的错误率,利用其中的 Variant Filtration算法进行过滤,过滤参数设置为:QUAL <30.0、QD <2.0、FS>60.0、MQ <40及cluster-Size-2-cluster-Window-Size 5[18]。最后使用SnpEff软件[19]对SNP和Small InDel进行注释。
1.4 变异基因的筛选与功能注释分析
基于大豆参考基因组的基础,统计非同义突变的SNP位点和编码区(CDS)发生 InDel变异的基因数量。基因功能通过序列比对,基于蛋白质直系同源簇(Clusters of Orthologous Groups of Proteins, COG)和京都基因与基金组百科全书(Kyoto Encyclopedia of Genes and Genomes, KEGG)数据库进行注释。
2. 结果与分析
2.1 测序数据质控评估
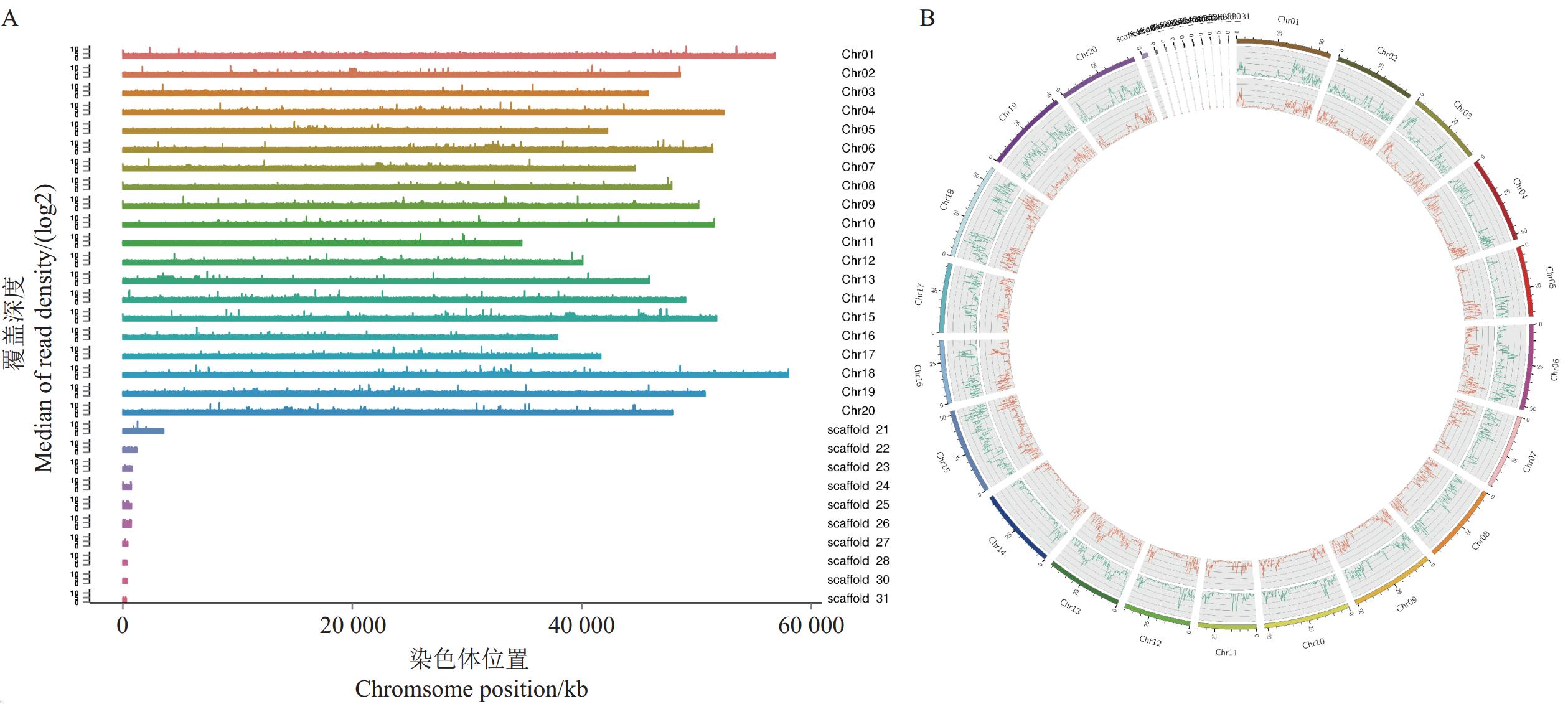
福豆234全基因组重测序数据通过合格质量控制,共获得
64799941 条Raw reads。Clean reads为剔除Raw reads中包含适配器的读数、ploy-N的读数和低质量读数,共得到64757037 条Clean reads。经过严格的数据质控和过滤后,Q30质控评分均值为91.82%,GC含量为35.90%。以大豆参考基因组Williams 82(Glyma.Wm82.a2v1, http://soybase.org)信息为参考,福豆234所得序列在大豆基因组中的定位比达98.66%,双端定位比达91.34%。测序覆盖深度达到17×,在1×的覆盖深度下,基因组上的覆盖度为98.08%;在5×的覆盖深度下,测序深度序列在基因组上的覆盖度达96.25%。福豆234染色体上的读数覆盖分布如图1A所示,读数覆盖更均匀地分布在染色体上。以上结果表明福豆234数据质量较高,序列能够在染色体上均匀分布,测序结果可信度高,可进行后续分析。变异检测结果表明,福豆234中共获得1478393 个SNP和356739 个Small InDel(图1B)。![]() 图 1 福豆234测序数据分析A:福豆234染色体覆盖深度分布;B:福豆234各类型变异在染色体的分布。Figure 1. Analysis of sequencing data of Fudou 234A: distribution of chromosomes coverage depth of Fudou 234; B: the distribution of the variations of Fudou 234 on the chromosomes.表 1 福豆234中与大豆蛋白质含量主要QTL区段候选基因SNP和Small Indel变异Table 1. SNP and Small InDel variations of candidate genes in main QTL segments related to soybean protein content in Fudou 234
图 1 福豆234测序数据分析A:福豆234染色体覆盖深度分布;B:福豆234各类型变异在染色体的分布。Figure 1. Analysis of sequencing data of Fudou 234A: distribution of chromosomes coverage depth of Fudou 234; B: the distribution of the variations of Fudou 234 on the chromosomes.表 1 福豆234中与大豆蛋白质含量主要QTL区段候选基因SNP和Small Indel变异Table 1. SNP and Small InDel variations of candidate genes in main QTL segments related to soybean protein content in Fudou 234基因名称
Gene ID变异类型 Variation type 基因名称
Gene ID变异类型 Variation type 基因名称
Gene ID变异类型 Variation type SNP Small InDel SNP Small InDel SNP Small InDel Glyma.20G082300 2 1 Glyma.20G086900 / 2 Glyma.10G133900 1 1 Glyma.20G082700 / 1 Glyma.20G087000 6 / Glyma.10G134100 1,7,2 7 Glyma.20G082800 7 / Glyma.20G087200 1 / Glyma.10G134400 1 1,7 Glyma.20G082900 2 / Glyma.15G048600 2 2 Glyma.10G134500 1,7 1 Glyma.20G083100 1,7,5,8,6,2 1 Glyma.15G048700 1, 7 2 Glyma.10G136100 1 / Glyma.20G083200 1,2 1,2 Glyma.15G048800 1, 7, 5,3, 2 1,11,7,3,2 Glyma.10G136300 2,1 2 Glyma.20G083300 1,7, 5,3,2 1,2 Glyma.15G048900 1, 4, 6,2 1,7,2 Glyma.10G136400 2 2 Glyma.20G083500 1,7,4,2 1,7,2 Glyma.15G049000 1,10,6,7,3,2 1,4,11,3,7 Glyma.10G136600 7 / Glyma.20G083600 1,10,5,6,3,4,2 4,2 Glyma.15G049500 3,5 / Glyma.10G136800 1,7,2 1,2,3 Glyma.20G083800 4,1 1 Glyma.15G049600 3,7,6,1 1,2,7 Glyma.08G182200 / 1 Glyma.20G084000 1,4,8,7,2 1,2 Glyma.15G049700 2,3,6,10,4,1 2,1 Glyma.08G182300 / 1,7 Glyma.20G084100 1,10,5,3,4,2 1,2,4 Glyma.15G049800 2,5,8,7,1 2,7,1 Glyma.08G182400 / 2 Glyma.20G084200 1,5 1 Glyma.15G049900 2,6,7,9,1 1,11,7,3,4 Glyma.08G182500 / 7 Glyma.20G084500 1,5,2 / Glyma.15G050100 2,7 2 Glyma.08G182700 / 1,2,7,9 Glyma.20G084900 1 / Glyma.15G050200 5,1 2,1 Glyma.08G182900 / 1,7 Glyma.20G085000 1 2 Glyma.15G050300 3 1 Glyma.08G183000 / 1,7,3,2 Glyma.20G085100 / 2 Glyma.15G050500 1,2,7 1,7 Glyma.08G183400 / 1 Glyma.20G085300 / 1 Glyma.15G050600 1,2,7 1 Glyma.08G183500 / 7,3,2 Glyma.20G085700 2 2 Glyma.10G132200 1,7,5,6,2 1,2 Glyma.08G183600 / 2 Glyma.20G085900 1 2 Glyma.10G132700 2,6 2,7 Glyma.08G183900 / 7,1 Glyma.20G086100 / 1 Glyma.10G132800 3,1 / Glyma.08G184100 / 2,7,1 Glyma.20G086800 2 2 Glyma.10G133700 2 / 11种变异类型中,1~11分别代表变异位点发生在基因上游区域、基因下游区域、基因的3’UTR内、基因的5’UTR内、编码区内同义突变、编码区内非同义突变、内含子、剪切位点区域、剪切受体突变、非编码区内起始密码子获得、密码子插入以及移码突变。
1~11 indicate mutation sites in 11 variation types occurred upstream, downstream, UTR 3 prime, UTR 5 prime, synonymous coding, non-synonymous coding, intron, splice site region, splice site donor, start gained, codon insertion, and frame shift, respectively.2.2 SNP检测与注释
重测序读数映射到大豆参考基因组Williams 82(Glyma.Wm82.a2v1, http://soybase.org),采用GATK软件进行对比,删除冗余SNP,福豆234共计
1478393 个高质量SNP,其中,转换类型的SNP(961071 个)多于颠换类型的SNP(517322 个),占比分别为65.01%和34.99%。转换/颠换比率(Ti/Tv)的平均值为1.85;纯合性的SNP较杂合性的SNP(233652 个)多,为1244741 个,占比为84.20%。总体杂合率为15.80%。福豆234基因组的SNP注释采用SnpEff软件进行。如图2所示,福豆234的SNP变异主要分布在基因间区,共鉴定出
673309 个(47.42%)。其次为基因上游区域(5000bp以内)、基因下游区域(5000bp以内)和内含子,分别为312129 (21.98%)、248175 (17.48%)和154082 个(10.88%)。此外,编码区域内的58618 个SNP中,获得了25318 个同义编码突变,占43.19%,以及32313 个非同义编码突变,占55.12%。![]() 图 2 福豆234的SNP注释结果A:福豆234的SNP变异分布全基因组注释结果;B:福豆234的SNP变异分布CDS注释结果。Figure 2. SNP annotations of Fudou 234A: genome-whole annotation statistics of SNP variant distribution of Fudou 234; B: CDS annotation statistics of SNP variant distribution of Fudou 234.
图 2 福豆234的SNP注释结果A:福豆234的SNP变异分布全基因组注释结果;B:福豆234的SNP变异分布CDS注释结果。Figure 2. SNP annotations of Fudou 234A: genome-whole annotation statistics of SNP variant distribution of Fudou 234; B: CDS annotation statistics of SNP variant distribution of Fudou 234.2.3 Small InDel检测与注释
福豆234重测序读数被映射到Williams 82参考基因组,去除重复的Small InDel,共鉴定出
356739 个Small InDel。其中,4965 个Small InDel变异从CDS区域鉴定得到。其中,插入、缺失、纯合突变和杂合突变从CDS区域鉴定得到,分别为2486 、2479 、3605 、1360 个。福豆234Small Indel长度在编码区域和全基因组的分布比较如图3所示。+l、−1、+3、−3类型变异在编码区域占较大比例。在全基因组范围内存在较多的+l、−1类型变异,而+3、−3类型变异较编码区域相对占比较小。![]() 图 3 福豆234的CDS区和全基因组Small Indel长度分布Figure 3. CDS region and distribution of Small Indel lengths in whole genome of Fudou 234
图 3 福豆234的CDS区和全基因组Small Indel长度分布Figure 3. CDS region and distribution of Small Indel lengths in whole genome of Fudou 234福豆234的Small InDel注释统计如图4所示。共鉴定出
111649 个Small InDel在全基因组中主要集中于基因间区(31.73%)。其次位于基因上游区域(5000 bp以内)(29.23%)、基因下游区域(5000 bp以内)(21.52%)和内含子(14.05%),分别为102833 、75716 、49430 个。共鉴定出2880 个位于编码区域的Small InDel主要集中于移码突变(45.67%)。其次为密码子的删除和插入、密码子插入和密码子删除。分别为1341 (21.27%)、724(11.48%)和617个(9.78%)。![]() 图 4 福豆234的Small InDel注释结果A:福豆234的Small InDel变异分布全基因组注释结果;B:福豆234的Small InDel变异分布CDS注释结果。Figure 4. Small InDel annotations of Fudou 234A: genome-whole annotation statistics of Small InDel variant distribution of Fudou 234; B: CDS annotation statistics of Small InDel variant distribution of Fudou 234.
图 4 福豆234的Small InDel注释结果A:福豆234的Small InDel变异分布全基因组注释结果;B:福豆234的Small InDel变异分布CDS注释结果。Figure 4. Small InDel annotations of Fudou 234A: genome-whole annotation statistics of Small InDel variant distribution of Fudou 234; B: CDS annotation statistics of Small InDel variant distribution of Fudou 234.2.4 遗传变异分析
基于大豆参考基因组Williams 82的基础上,福豆234基因组一共鉴定出
18509 个突变基因。其中,共鉴定出14323 个非同义SNP突变的基因,占77.38%;共鉴定出4186 个Small Indel的突变基因位于CDS区域,占22.62%。将福豆234基于DNA水平变异进行COG和KEGG数据库进行注释。基于COG注释结果(图5),信号传导机制(684个),转录(559个),碳水化合物转输和代谢(555个)等在福豆234存在大量的变异基因。根据福豆234基因组重测序进行KEGG注释如图6所示。变异基因主要注释到KEGG代谢通路涉及碳代谢(ko 01200)156个、淀粉和蔗糖代谢(ko 00500)147个,氨基酸生物合成(ko 01230)142个,植物激素信号传导(ko 04075)137个,内质网蛋白质加工(ko 04141)110个。
前人研究表明20号染色体(I连锁群,29.8-31.6 Mbp)和15号染色体(E连锁群,38.1-39.7 Mbp)在自然环境和复杂的遗传种群中定位到大豆籽粒蛋白质含量QTL[20−21]。Paril等[22]对两个主要的大豆蛋白质含量QTL(Chr.20和15)以及区域之间的片段复制区间预测出86个候选基因。本研究对福豆234基于全基因组测序基础上,根据之前报道的候选基因进行分析。由表1所示,共鉴定出65个候选基因存在SNP或Small InDel变异。其中SNP变异类型达10种,占比为15.38%,主要集中于基因上游区域和基因下游区域。其次是内含子区域和编码区内同义突变,其他变异类型较少,无密码子插入和移码突变,CDS区域内非同义突变有16个位点,涉及10个基因;Small InDel变异类型相对SNP变异类型而言相对较少,仅7种,占比为10.77%。主要集中在基因上游区域和基因下游区域,其次是内含子区域,其他变异类型较少。
3. 讨论
大豆是食用和饲用植物蛋白的主要来源,粒用大豆含有38%~42%的蛋白质[22]。随着全球消费者对大豆蛋白质需求日益增加,这一比例还在逐年上升[23]。因此,通过遗传改良选育高蛋白大豆品种是育种工作者的目标之一。大豆作为豆科植物的一个典型,自花授粉特性对保持基因组同质性和减少基因组变异产生强烈影响。由于大豆基因组的复杂性,挖掘高蛋白含量的优异特性也具有一定的局限性。自大豆种质资源全基因组测序的完成,大豆基因组遗传图谱揭示加快了新一代大豆分子育种进程[24−25]。本研究通过全基因组重测序技术,比较于Williams 82参考基因组,在高蛋白大豆福豆234共获得
1478393 个SNP和356739 个Small InDel,变异产生的突变基因有18509 个。变异基因COG注释主要发生在信号传导机制类、转录类、碳水化合物转输和代谢类等途径,而在KEGG注释主要发生在碳代谢、淀粉和蔗糖代谢、氨基酸生物合成、植物激素信号传导以及内质网蛋白加工等途径。大豆籽粒中含有40%蛋白质,为脂肪(20%)的两倍,虽然蛋白质单位生物能低于脂肪,但在物质分配和能量转运占据优势[26]。籽粒蛋白质积累与蔗糖合成酶活性呈正比,其活性影响蔗糖卸载量进而影响碳代谢[27]。本研究根据已报道的与大豆籽粒蛋白质含量相关的QTL通过共线性分析预测到的86个候选基因,对福豆234的变异基因进行SNP和Small InDel分析,与参考基因组Williams 82相比,共有65个候选基因发生SNP或Small InDel变异,其中有10个候选基因编码区内至少一个位点发生非同义突变。王嘉等[28]对大豆籽粒蛋白质全基因组重测序及QTL定位。共获得
2453344 个SNP和476953 个Small Indel,变异产生的突变基因共38896 个,14491 个基因存在2种或2种以上类型的突变。其中,候选基因Gy5(Glyma.13G123500)、ABI3(Glyma.18G176100)和CAR1(Glyma.03G163500)与大豆储藏蛋白合成相关。此外,QTL结果表明,与大豆籽粒蛋白质含量相关的6个QTL位点分别分布在 C2、E、I和M4连锁群上。此外,Fliege等[29]研究发现,位于20号染色体的Glyma.20G085100基因影响大豆籽粒蛋白质含量,在低蛋白大豆导入该基因有助于提高2%的蛋白质含量。糖作为信号分子参与植物生长发育,其中糖转运蛋白是植物体运输途径的关键组成部分,包括单糖转运蛋白、蔗糖转运蛋白和糖最终输出转运蛋白(Sugar will eventually be exported transporter,SWEET)[30−31]。蔗糖在成熟叶片叶肉细胞中合成,SWEET蛋白为韧皮部转载之前介导蔗糖从叶肉细胞流出到质外体的核心参与者[32]。另一方面,SWEET蛋白与其他蛋白质相互协调,调节“源-流-库”关系[33]。大豆籽粒由成熟胚组成,胚胎发育时期,微小的胚胎快速生长并从发育中种子的胚乳中获取大量糖分[34]。之前的研究表明GmSWEET24(Glyma.08G183500)在籽粒发育时期表达量较高[35−36]。WAT1(Walls are thin 1)是一种细胞内生长素转运蛋白,对木质部前体中生长素积累有直接作用[37]。Pal等[38]研究发现参与植物激素信号传导的WAT1对油菜种子重量和产量具有显著影响。Liu等[39]研究表明3个编码WAT1相关蛋白不仅与苎麻纤维产量呈高度相关,同时发现与氮代谢相关正向选择基因共计22个,证实与参与粗蛋白含量和营养生长等驯化性状。本研究结果表明,候选基因Glyma.15G049700、Glyma.15G049800和Glyma.15G049900具有WAT1的功能,可能是促进福豆234高产和高蛋白特征的关键。4. 结论
大豆籽粒蛋白质含量是典型的数量性状,一般是由主基因控制,还受到微效多基因和环境因素的影响,不同大豆蛋白质含量基因间存在独立或互作的关系[40]。本研究对高蛋白大豆品种福豆234全基因组重测序数据分析,获得
1478393 个SNP,356739 个Small InDel。通过COG分析发现信号传导机制、转录、碳水化合物转输和代谢等和KEGG分析发现碳代谢、淀粉和蔗糖代谢、氨基酸生物合成、植物激素信号传导、内质网蛋白质加工等通路与福豆234遗传变异相关。由变异产生的突变基因达18509 个,从先前报道过的15号染色体和20号染色体区段内的基因进行变异分析,65个基因发生SNP或Small InDel水平的变异,发现SNP变异类型占主体,而Small InDel变异类型相对较少。本研究初步阐明了福豆234全基因组突变类型,为大豆高蛋白品种的选育和优异基因挖掘奠定基础,可进一步为基因研究和分子标记辅助育种等提供大量的分子标记资源,为研究该性状的遗传调控提供方向。 -
![]()
图 1 福豆234测序数据分析
A:福豆234染色体覆盖深度分布;B:福豆234各类型变异在染色体的分布。
Figure 1. Analysis of sequencing data of Fudou 234
A: distribution of chromosomes coverage depth of Fudou 234; B: the distribution of the variations of Fudou 234 on the chromosomes.
![]()
图 2 福豆234的SNP注释结果
A:福豆234的SNP变异分布全基因组注释结果;B:福豆234的SNP变异分布CDS注释结果。
Figure 2. SNP annotations of Fudou 234
A: genome-whole annotation statistics of SNP variant distribution of Fudou 234; B: CDS annotation statistics of SNP variant distribution of Fudou 234.
![]()
图 3 福豆234的CDS区和全基因组Small Indel长度分布
Figure 3. CDS region and distribution of Small Indel lengths in whole genome of Fudou 234
![]()
图 4 福豆234的Small InDel注释结果
A:福豆234的Small InDel变异分布全基因组注释结果;B:福豆234的Small InDel变异分布CDS注释结果。
Figure 4. Small InDel annotations of Fudou 234
A: genome-whole annotation statistics of Small InDel variant distribution of Fudou 234; B: CDS annotation statistics of Small InDel variant distribution of Fudou 234.
表 1 福豆234中与大豆蛋白质含量主要QTL区段候选基因SNP和Small Indel变异
Table 1 SNP and Small InDel variations of candidate genes in main QTL segments related to soybean protein content in Fudou 234
基因名称
Gene ID变异类型 Variation type 基因名称
Gene ID变异类型 Variation type 基因名称
Gene ID变异类型 Variation type SNP Small InDel SNP Small InDel SNP Small InDel Glyma.20G082300 2 1 Glyma.20G086900 / 2 Glyma.10G133900 1 1 Glyma.20G082700 / 1 Glyma.20G087000 6 / Glyma.10G134100 1,7,2 7 Glyma.20G082800 7 / Glyma.20G087200 1 / Glyma.10G134400 1 1,7 Glyma.20G082900 2 / Glyma.15G048600 2 2 Glyma.10G134500 1,7 1 Glyma.20G083100 1,7,5,8,6,2 1 Glyma.15G048700 1, 7 2 Glyma.10G136100 1 / Glyma.20G083200 1,2 1,2 Glyma.15G048800 1, 7, 5,3, 2 1,11,7,3,2 Glyma.10G136300 2,1 2 Glyma.20G083300 1,7, 5,3,2 1,2 Glyma.15G048900 1, 4, 6,2 1,7,2 Glyma.10G136400 2 2 Glyma.20G083500 1,7,4,2 1,7,2 Glyma.15G049000 1,10,6,7,3,2 1,4,11,3,7 Glyma.10G136600 7 / Glyma.20G083600 1,10,5,6,3,4,2 4,2 Glyma.15G049500 3,5 / Glyma.10G136800 1,7,2 1,2,3 Glyma.20G083800 4,1 1 Glyma.15G049600 3,7,6,1 1,2,7 Glyma.08G182200 / 1 Glyma.20G084000 1,4,8,7,2 1,2 Glyma.15G049700 2,3,6,10,4,1 2,1 Glyma.08G182300 / 1,7 Glyma.20G084100 1,10,5,3,4,2 1,2,4 Glyma.15G049800 2,5,8,7,1 2,7,1 Glyma.08G182400 / 2 Glyma.20G084200 1,5 1 Glyma.15G049900 2,6,7,9,1 1,11,7,3,4 Glyma.08G182500 / 7 Glyma.20G084500 1,5,2 / Glyma.15G050100 2,7 2 Glyma.08G182700 / 1,2,7,9 Glyma.20G084900 1 / Glyma.15G050200 5,1 2,1 Glyma.08G182900 / 1,7 Glyma.20G085000 1 2 Glyma.15G050300 3 1 Glyma.08G183000 / 1,7,3,2 Glyma.20G085100 / 2 Glyma.15G050500 1,2,7 1,7 Glyma.08G183400 / 1 Glyma.20G085300 / 1 Glyma.15G050600 1,2,7 1 Glyma.08G183500 / 7,3,2 Glyma.20G085700 2 2 Glyma.10G132200 1,7,5,6,2 1,2 Glyma.08G183600 / 2 Glyma.20G085900 1 2 Glyma.10G132700 2,6 2,7 Glyma.08G183900 / 7,1 Glyma.20G086100 / 1 Glyma.10G132800 3,1 / Glyma.08G184100 / 2,7,1 Glyma.20G086800 2 2 Glyma.10G133700 2 / 11种变异类型中,1~11分别代表变异位点发生在基因上游区域、基因下游区域、基因的3’UTR内、基因的5’UTR内、编码区内同义突变、编码区内非同义突变、内含子、剪切位点区域、剪切受体突变、非编码区内起始密码子获得、密码子插入以及移码突变。
1~11 indicate mutation sites in 11 variation types occurred upstream, downstream, UTR 3 prime, UTR 5 prime, synonymous coding, non-synonymous coding, intron, splice site region, splice site donor, start gained, codon insertion, and frame shift, respectively. 下载: 导出CSV
下载: 导出CSV
-
[1] CAO P, ZHAO Y, WU F J, et al. Multi-omics techniques for soybean molecular breeding [J]. International Journal of Molecular Sciences, 2022, 23(9): 4994. DOI: 10.3390/ijms23094994
[2] XU X Y, BAI G H. Whole-genome resequencing: Changing the paradigms of SNP detection, molecular mapping and gene discovery [J]. Molecular Breeding, 2015, 35(1): 33. DOI: 10.1007/s11032-015-0240-6
[3] HUANG X H, FENG Q, QIAN Q, et al. High-throughput genotyping by whole-genome resequencing [J]. Genome Research, 2009, 19(6): 1068−1076. DOI: 10.1101/gr.089516.108
[4] PETEREIT J, MARSH J I, BAYER P E, et al. Genetic and genomic resources for soybean breeding research [J]. Plants, 2022, 11(9): 1181. DOI: 10.3390/plants11091181
[5] YANG C M, YAN J, JIANG S Q, et al. Resequencing 250 soybean accessions: New insights into genes associated with agronomic traits and genetic networks[J]. Genomics, Proteomics & Bioinformatics, 2022, 20(1): 29-41.
[6] LIU N, NIU Y C, ZHANG G W, et al. Genome sequencing and population resequencing provide insights into the genetic basis of domestication and diversity of vegetable soybean [J]. Horticulture Research, 2022, 9: uhab052. DOI: 10.1093/hr/uhab052
[7] LEE K J, KIM D S, KIM J B, et al. Identification of candidate genes for an early-maturing soybean mutant by genome resequencing analysis [J]. Molecular Genetics and Genomics, 2016, 291(4): 1561−1571. DOI: 10.1007/s00438-016-1183-2
[8] MALDONADO DOS SANTOS J V, VALLIYODAN B, JOSHI T, et al. Evaluation of genetic variation among Brazilian soybean cultivars through genome resequencing [J]. BMC Genomics, 2016, 17: 110. DOI: 10.1186/s12864-016-2431-x
[9] JIANG H, JIA H Y, HAO X S, et al. Mapping Locus RSC11K and predicting candidate gene resistant to Soybean mosaic virus strain SC11 through linkage analysis combined with genome resequencing of the parents in soybean [J]. Genomics, 2022, 114(4): 110387. DOI: 10.1016/j.ygeno.2022.110387
[10] YUAN Y, YANG Y Q, SHEN Y C, et al. Mapping and functional analysis of candidate genes involved in resistance to soybean (Glycine max) mosaic virus strain SC3 [J]. Plant Breeding, 2020, 139(3): 618−625. DOI: 10.1111/pbr.12799
[11] 林国强, 张轼, 滕振勇, 等. 高蛋白大豆福豆234的选育及高产农艺措施数学模型 [J]. 福建农业学报, 2005, 20(2):69−73. DOI: 10.3969/j.issn.1008-0384.2005.02.002 LIN G Q, ZHANG S, TENG Z Y, et al. Breeding and mathematical model of agronomic measures for high yield and protein content soybean variety Fudou 234 [J]. Fujian Journal of Agricultural Sciences, 2005, 20(2): 69−73. (in Chinese) DOI: 10.3969/j.issn.1008-0384.2005.02.002
[12] ABOUL-MAATY N A F, ORABY H A S. Extraction of high-quality genomic DNA from different plant orders applying a modified CTAB-based method [J]. Bulletin of the National Research Centre, 2019, 43(1): 25. DOI: 10.1186/s42269-019-0066-1
[13] 张彦威, 李伟, 张礼凤, 等. 基于重测序的大豆新品种齐黄34的全基因组变异挖掘 [J]. 中国油料作物学报, 2016, 38(2):150−158. DOI: 10.7505/j.issn.1007-9084.2016.02.003 ZHANG Y W, LI W, ZHANG L F, et al. Genome-wide variations of soybean cultivar Qihuang 34 by whole genome re-sequencing [J]. Chinese Journal of Oil Crop Sciences, 2016, 38(2): 150−158. (in Chinese) DOI: 10.7505/j.issn.1007-9084.2016.02.003
[14] 郭丹丹, 袁凤杰, 郁晓敏. 基于重测序的籽粒型和鲜食型大豆的全基因组变异分析 [J]. 分子植物育种, 2019, 17(22):7306−7312. GUO D D, YUAN F J, YU X M. Genome-wide variation analysis of grain and vegetable soybeans based on re-sequencing [J]. Molecular Plant Breeding, 2019, 17(22): 7306−7312. (in Chinese)
[15] LI H, DURBIN R. Fast and accurate short read alignment with Burrows-Wheeler transform [J]. Bioinformatics, 2009, 25(14): 1754−1760. DOI: 10.1093/bioinformatics/btp324
[16] LI H, HANDSAKER B, WYSOKER A, et al. The Sequence Alignment/Map format and SAMtools [J]. Bioinformatics, 2009, 25(16): 2078−2079. DOI: 10.1093/bioinformatics/btp352
[17] MCKENNA A, HANNA M, BANKS E, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data [J]. Genome Research, 2010, 20(9): 1297−1303. DOI: 10.1101/gr.107524.110
[18] 沈丽丽, 曹斌斌, 杨光耀, 等. 毛竹及其2种竿型变异类型的全基因组重测序分析 [J]. 基因组学与应用生物学, 2023, 42(6):581−592. SHEN L L, CAO B B, YANG G Y, et al. Whole genome resequencing analysis of moso bamboo(Phyllostachys edulis)and its two culm variants [J]. Genomics and Applied Biology, 2023, 42(6): 581−592. (in Chinese)
[19] CINGOLANI P, PLATTS A, WANG L L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3 [J]. Fly, 2012, 6(2): 80−92. DOI: 10.4161/fly.19695
[20] 魏荷, 王金社, 卢为国. 大豆籽粒蛋白质含量分子遗传研究进展 [J]. 中国油料作物学报, 2015, 37(3):394−410. DOI: 10.7505/j.issn.1007-9084.2015.03.021 WEI H, WANG J S, LU W G. Molecular genetic advances in soybean seed protein [J]. Chinese Journal of Oil Crop Sciences, 2015, 37(3): 394−410. (in Chinese) DOI: 10.7505/j.issn.1007-9084.2015.03.021
[21] BANDILLO N, JARQUIN D, SONG Q J, et al. A population structure and genome-wide association analysis on the USDA soybean germplasm collection[J]. The Plant Genome, 2015, 8(3): eplantgenome2015.04. 0024.
[22] PATIL G, MIAN R, VUONG T, et al. Molecular mapping and genomics of soybean seed protein: A review and perspective for the future [J]. Theoretical and Applied Genetics, 2017, 130(10): 1975−1991. DOI: 10.1007/s00122-017-2955-8
[23] VAN K, MCHALE L K. Meta-analyses of QTLs associated with protein and oil contents and compositions in soybean[Glycine max (L.) Merr. ]seed [J]. International Journal of Molecular Sciences, 2017, 18(6): 1180. DOI: 10.3390/ijms18061180
[24] LAM H M, XU X, LIU X, et al. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection [J]. Nature Genetics, 2010, 42: 1053−1059. DOI: 10.1038/ng.715
[25] SCHMUTZ J, CANNON S B, SCHLUETER J, et al. Genome sequence of the palaeopolyploid soybean [J]. Nature, 2010, 463: 178−183. DOI: 10.1038/nature08670
[26] 郭茜茜. 大豆子粒蛋白质积累与碳代谢关系的研究[D]. 哈尔滨: 东北农业大学, 2010:16-41. GUO Q Q. Study on the relationship between protein accumulation and carbon metabolism in soybean seeds[D]. Harbin: Northeast Agricultural University, 2010:16-41. (in Chinese)
[27] PATIL G, VUONG T D, KALE S, et al. Dissecting genomic hotspots underlying seed protein, oil, and sucrose content in an interspecific mapping population of soybean using high-density linkage mapping [J]. Plant Biotechnology Journal, 2018, 16(11): 1939−1953. DOI: 10.1111/pbi.12929
[28] 王嘉, 曾召琼, 梁建秋, 等. 基于全基因组重测序的大豆分子标记开发及籽粒蛋白质含量QTL定位 [J]. 中国农业科学, 2019, 52(16):2743−2757. DOI: 10.3864/j.issn.0578-1752.2019.16.001 WANG J, ZENG Z Q, LIANG J Q, et al. Development new molecular markers for quantitative trait locus (QTL) analysis of the seed protein content based on whole genome re-sequencing in soybean [J]. Scientia Agricultura Sinica, 2019, 52(16): 2743−2757. (in Chinese) DOI: 10.3864/j.issn.0578-1752.2019.16.001
[29] FLIEGE C E, WARD R A, VOGEL P, et al. Fine mapping and cloning of the major seed protein quantitative trait loci on soybean chromosome 20 [J]. The Plant Journal: for Cell and Molecular Biology, 2022, 110(1): 114−128. DOI: 10.1111/tpj.15658
[30] MA Q J, SUN M H, LU J, et al. Transcription factor AREB2 is involved in soluble sugar accumulation by activating sugar transporter and amylase genes [J]. Plant Physiology, 2017, 174(4): 2348−2362. DOI: 10.1104/pp.17.00502
[31] 张计育, 王刚, 王涛, 等. SWEET蛋白在植物生长发育中的功能作用研究进展[J]. 植物资源与环境学报, 2023, 32(5): 1-15. ZHANG J Y, WANG G, WANG T, et al. Research progress on functional roles of SWEET proteins in plant growth and development[J]. Journal of Plant Resources and Environment, 2023, 32(5): 1-15. (in Chinese) Development[J]. Journal of Plant Resources and Environment, 2023, 32(5): 1-15. (in Chinese)
[32] CHEN L Q, QU X Q, HOU B H, et al. Sucrose efflux mediated by SWEET proteins as a key step for phloem transport [J]. Science, 2012, 335(6065): 207−211. DOI: 10.1126/science.1213351
[33] TAKAHASHI F, SATO-NARA K, KOBAYASHI K, et al. Sugar-induced adventitious roots in Arabidopsis seedlings [J]. Journal of Plant Research, 2003, 116(2): 83−91. DOI: 10.1007/s10265-002-0074-2
[34] WANG S D, YOKOSHO K, GUO R Z, et al. The soybean sugar transporter GmSWEET15 mediates sucrose export from endosperm to early embryo [J]. Plant Physiology, 2019, 180(4): 2133−2141. DOI: 10.1104/pp.19.00641
[35] 柯博洋, 李文龙, 张彩英. 大豆SWEET基因在荚粒发育过程中与逆境胁迫下的表达 [J]. 中国农业科技导报, 2023, 25(8):33−52. KE B Y, LI W L, ZHANG C Y. Expressions of SWEET genes during pod and seed developments and under different stress conditions in soybean [J]. Journal of Agricultural Science and Technology, 2023, 25(8): 33−52. (in Chinese)
[36] PATIL G, VALLIYODAN B, DESHMUKH R, et al. Soybean (Glycine max) SWEET gene family: Insights through comparative genomics, transcriptome profiling and whole genome re-sequence analysis [J]. BMC Genomics, 2015, 16(1): 520. DOI: 10.1186/s12864-015-1730-y
[37] RANOCHA P, DENANCÉ N, VANHOLME R, et al. Walls are thin 1 (WAT1), an Arabidopsis homolog of Medicago truncatula NODULIN21, is a tonoplast-localized protein required for secondary wall formation in fibers [J]. The Plant Journal: for Cell and Molecular Biology, 2010, 63(3): 469−483. DOI: 10.1111/j.1365-313X.2010.04256.x
[38] PAL L, SANDHU S K, BHATIA D, et al. Genome-wide association study for candidate genes controlling seed yield and its components in rapeseed (Brassica napus subsp. napus) [J]. Physiology and Molecular Biology of Plants, 2021, 27(9): 1933−1951. DOI: 10.1007/s12298-021-01060-9
[39] LIU C, ZENG L B, ZHU S Y, et al. Draft genome analysis provides insights into the fiber yield, crude protein biosynthesis, and vegetative growth of domesticated ramie (Boehmeria nivea L. Gaud) [J]. DNA Research: an International Journal for Rapid Publication of Reports on Genes and Genomes, 2018, 25(2): 173−181. DOI: 10.1093/dnares/dsx047
[40] 刘顺湖, 周瑞宝, 盖钧镒. 大豆蛋白质有关性状遗传的分离分析 [J]. 作物学报, 2009, 35(11):1958−1966. DOI: 10.3724/SP.J.1006.2009.01958 LIU S H, ZHOU R B, GAI J Y. Segregation analysis for inheritance of protein related traits in soybean[Glycine max (L. ) merr. [J]. Acta Agronomica Sinica, 2009, 35(11): 1958−1966. (in Chinese) DOI: 10.3724/SP.J.1006.2009.01958





计量
- 文章访问数: 209
- HTML全文浏览量: 101
- PDF下载量: 56
